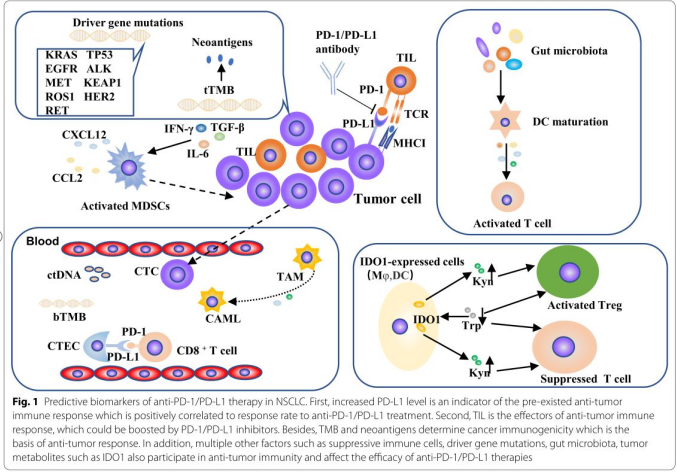
肿瘤突变量(TMB)
采用全外显子组测序(WES)和癌症基因面板测序(CGPs)对肿瘤组织中的脱氧核糖核酸(DNA)突变进行测量[28]。肿瘤组织TMB (tTMB)与肿瘤新抗原负荷呈正相关(图1)[29]。多项回顾性研究表明,tTMB与PD-1/PD-L1抑制剂的疗效及患者预后密切相关。在KEYNOTE-158中,对于接受派姆单抗治疗的患者,tTMBHigh组比非tTMBHigh组有更高的客观缓解率(29 vs. 6%)(表1)[30]。在CHECKMATE-026研究中,接受nivolumab治疗的高tTMB患者的无进展生存期(PFS)更长(9.7个月vs. 5.8个月;HR = 0.62, 95% CI: 0.38 - 1.00)(表1),且有效率(47 vs. 28%)高于接受化疗[31]的患者。同样,CHECKMATE-227的结果显示,在tTMB高的患者中,nivolumab + ipilimumab组的PFS比化疗组更长(7.2个月vs. 5.5个月;HR = 0.58, 97.5% CI: 0.41-0.81, p < 0.001)[32](表1)。血液TMB (bTMB)被发现是一种新的微创选择,它通过检测血浆细胞游离DNA(图1)。bTMB与ttmb[28]呈正相关。与bTMB < 6亚组相比,bTMB≥6亚组有更高的客观有效率(39.3 vs. 9.1%)和更长的PFS (HR = 0.39, 95% CI: 0.18-0.84, p = 0.01)(表1)然而,bTMB与患者生存期之间呈非线性相关。对于使用PD-L1抑制剂的患者,bTMB-High(≥14个突变/Mb)和bTMB-Low(≤7个突变/Mb)亚组比bTMB-Medium(8-13个突变/Mb)亚组的PFS和OS更长。基线循环肿瘤DNA (ctDNA)与bTMB评分呈正相关解释了bTMBlow患者预后较好。此外,与bTMB-Medium患者相比,bTMB-low患者有更长的反应持续时间和更高的稳定患病率[33]。总之,高突变促进了肿瘤新抗原的产生,增强了肿瘤免疫原性,提高了对PD-L1抑制剂[34]的应答率。
新抗原
新抗原来源于体细胞突变[35],与主要组织相容性I类(MHCI)结合并在癌细胞表面表达。新抗原赋予肿瘤高免疫原性并诱导抗肿瘤免疫应答(图1)[36]。肿瘤细胞释放新抗原并被专业APC捕获,然后靶向肿瘤特异性抗原的效应T细胞被激活[37]。活化的T细胞迁移并浸润到肿瘤床,特异性地识别肿瘤细胞上的抗原并杀死癌细胞[37]。免疫原性强的肿瘤克隆被清除,免疫原性弱的癌细胞逃脱免疫监视[38]。许多研究证明,抗pd -1/PD-L1联合放疗或溶瘤病毒可增加新抗原的释放,增强特异性免疫应答[39-41]。与无持续临床获益(NDB)的患者相比,DCB患者有更高的候选新抗原负担。高候选新抗原负荷与PFS改善相关(HR = 0.23, 95%CI: 0.09-0.58, p = 0.002)[42](表1)。免疫治疗的疗效不仅与新抗原的数量有关,还与新抗原的质量有关[43]。高质量的新抗原尤其是克隆型新抗原可以与多种HLA等位基因结合[43]。克隆性新抗原促进了高表达PD-1的新抗原反应T细胞的活化和浸润,而肿瘤富集的克隆性新抗原对PD-1阻滞剂更敏感[34]。DCB的发生率高突变患者负担和低neoantigen subclonal分数高于患者高subclonal neoantigen分数或低克隆neoantigen负担(11% vs 92)[34](表1)。免疫消除neoantigen-containing肿瘤细胞亚群和遗传染色体等事件肿瘤细胞中杂合性缺失或丢失导致新抗原的丢失,从而导致获得性抗pd -1/PD-L1治疗的产生[44]。
驱动基因突变
新一代测序(NGS)被广泛用于肿瘤基因组分析[45]。通过靶向NGS检测到的基因改变可能预示着PD-1/PD-L1抑制剂的应答率(图1)[45]。Kirsten大鼠肉瘤2病毒癌基因同源基因(KRAS)突变状态与PD-L1表达呈正相关[46]。此外,KRAS突变型肿瘤具有更多的TILs和更高的TMB,表现为适应性免疫抵抗的炎症表型和免疫原性增加[46]。与KRAS野生亚组相比,KRAS突变亚组有更高的客观缓解率(比值比= 1.51,95% CI: 1.17-1.96, p = 0.002)(表1)tp53突变的肿瘤具有高PD-L1表达和CD8+ T细胞密度[47]。TP53突变且无STK11(丝氨酸/苏氨酸激酶11)或表皮生长因子受体(EGFR)共突变的患者抗pd -1治疗有效率更高,PFS更长(HR = 0.32, 95%CI: 0.16-0.63, p < 0.001)。与免疫细胞毒性、T细胞趋化、抗原处理相关的通路在这种肿瘤亚型中上调[47]。19外显子缺失、L858R突变和T790M突变的EGFR上调PD-L1的表达,从而减弱淋巴细胞的细胞毒性,通过PD-1/PD-L1轴诱导t细胞衰竭[48-50]。在接受抗pd -1治疗的患者中,EGFR突变的患者预后更差(中位PFS: 5.3个月,95% CI: 1.3-12.4个月)[51]。间变性淋巴瘤激酶(ALK)重组上调PD-L1表达,促进肿瘤免疫逃逸[52]。然而,接受抗pd -1治疗的ALK突变患者比EGFR突变患者的PFS更差(ALK: 0.6 (95% CI: 0.2-2.1)个月,EGFR: 1.8 (95% CI: 1.2-2.1)个月),提示PD-L1表达并不是ALK重排[53]患者免疫治疗的可靠生物标志物。间充质上皮细胞转化(MET)外显子14跳跃性改变发生在3%-4%的肺癌中[54]。大部分MET外显子14改变的肺癌细胞表达PD-L1[55]。MET外显子14突变的肺癌患者对单药或联合免疫检查点抑制剂(客观有效率:17%,95%置信区间:6%-36%)反应温和[55],似乎没有从免疫治疗中获益[56]。Kelch-like ECH-associated protein 1 (KEAP1)体细胞突变通过激活KEAP1/核因子红系-2相关因子-2 (nuclear factor红系-2-related factor-2, NFE2L2)应激反应通路促进肿瘤发生,降低治疗敏感性[57-60]。NFE2L2/KEAP1突变与高TMB和PD-L1表达相关,免疫治疗NFE2L2/KEAP1突变患者的疗效优于其他治疗(中位OS: 22.52个月vs. 12.89个月,p = 0.0034)[61]。其他稀有驱动基因如ROS1、HER2、RET的突变状态也可能影响对PD-1/PD-L1抑制剂的应答[62,63]。
炎症相关基因
一些表达特征反映了肿瘤的炎症状态,如与T细胞活化、趋化因子表达和适应性免疫抵抗相关的基因(图1)[64,65]。炎症评分显著升高的患者往往对PD-1/PD-L1抑制剂敏感。与无应答者相比,应答者的炎症特征评分显著高于无应答者[65]。此外,炎症评分与上皮-间质转化(EMT)评分相关。Thompson的研究表明,结合EMT表型特征评分和炎症基因评分提高了预测的准确性[65]。因此,可以预测,逆转EMT可能提高抗pd -1/PD-L1治疗的耐药性[65]。进一步研究发现,在同一NSCLC队列中,与抗原加工机械(APM)评分相关的8个基因比炎症评分更能有效预测疗效[66]。此外,我们之前的研究表明,一些免疫反应相关的特征与免疫检查点抑制剂在肺腺癌中的疗效相关[4]。
microRNA(miRNA)
MiRNA通过调节蛋白翻译来修饰靶基因的表达[67]。miRNA失调与癌变密切相关,可通过靶向一组基因促进或抑制癌症(图1)[68]。此外,miRNA调节抗肿瘤免疫。一些miRNAs干扰抗原的加工和递呈,上调人白细胞抗原(HLA)-G表达,下调自然杀伤组2,成员D (NKG2D)配体形成免疫逃逸[69]。环状RNA circ-CPA4通过下调let-7 miRNA上调NSCLC细胞中PD-L1的表达[70]。10个高表达mirna (miR-93、miR-138-5p、miR-200、miR-27a、miR-424、miR-34a、miR28、miR-106b、miR-193a-3p、miR-181a)在抗pd -1治疗的应答者中发现,并与明显改善的PFS和OS相关(中位PFS: 6.25个月vs. 3.21个月,HR = 0.45, 95% CI: 0.25-0.76;中位OS: 7.65个月vs. 3.2个月,HR = 0.39, 95% CI: 0.15-0.68)(表1)[71]。
肿瘤微环境相关生物标志物
肿瘤浸润淋巴细胞(TIL)
既往研究表明PD-L1表达与非小细胞肺癌肿瘤内T细胞浸润显著相关[72]。转录因子的胸腺细胞selection-associated高机动组框基因(托克斯)在肿瘤浸润CD8 + T细胞促进T细胞疲惫的表达上调免疫蛋白质PD-1检查站,T细胞免疫球蛋白和mucin-domain包含3 (TIM-3) [73], T细胞免疫球蛋白和ITIM域(TIGIT) [74],细胞毒性T淋巴细胞抗原4 (CTLA-4),从而降低抗pd -1治疗的结果(图1)[75]。根据PD-L1/TIL状态,NSCLC肿瘤免疫微环境分为I型(PD-L1+, TIL+)、II型(PD-L1−,TIL−)、III型(PD-L1+, TIL−)和IV型(PD-L1−,TIL+)[76]。不同肿瘤免疫微环境类型相关的临床因素的差异决定了患者对联合免疫治疗的选择[76]。抗pd -1/PD-L1治疗对I型肿瘤有很大的益处。然而,III型肿瘤对anti-PD-1/PD-L1单药治疗具有耐药性,这可以通过联合辅助治疗将T细胞引入肿瘤床来逆转[77]。CD8+细胞在CD3+ til总人群中的比例与抗pd -1/ PD-L1治疗结果密切相关。研究表明,高cd8 / cd3比值与无病生存(DFS)和OS呈正相关(DFS: HR = 0.954, 95%CI: 0.965-0.983, p = 0.002;OS: HR = 0.965, 95%CI: 0.931 - 1.001, p = 0.057)(表1)[78]。抗pd -1治疗后CD8+ T细胞的早期增殖预示着抗pd -1治疗的良好临床反应[79]。T细胞受体(T cell receptor, TCR)表达于T细胞表面,由α链和β链组成,通过体细胞DNA重排形成多样性和特异性[80]。TCR与MHC/抗原短肽复合物结合并引发免疫应答(图1)[81]。采用多重PCR技术对PD-1+ CD8+ T细胞的TCR β链互补区3进行测序。TCR anti-PD-1 / PD-L1治疗前的多样性预示着更好的生存结果(6.4和2.5个月,HR = 0.39, 95%置信区间CI: 0.17 - -0.94, p = 0.021),治疗后细胞的单克隆也预示临床效益(7.3和2.6个月,HR = 0.26, 95%置信区间CI: 0.08 - -0.86, p = 0.002)[82]。与安慰剂组相比,同期放化疗(cCRT)后使用durvalumab进行巩固治疗可显著提高患者的总生存期和中位无进展生存期[83]。放射治疗通过促进肿瘤新抗原的释放和驱动CD8 + TILs的免疫攻击来激发抗肿瘤免疫[84]。cCRT后PD-L1的上调可能是对放疗相关免疫攻击的反应,这为PD-L1阻遏剂在cCRT后的应用提供了理论依据[85]。此外,cCRT后CD8 + TIL密度增加与良好的生存率相关[85]。
抑制免疫细胞
T肿瘤浸润调节性T淋巴细胞(Tregs)表面表达PD-L1、PD-L2,高度抑制肿瘤特异性效应T细胞的活性[86]。吲哚胺2,3-双加氧酶1 (IDO1)通过肿瘤微环境中l-色氨酸(Trp)耗竭和kynurenine (Kyn)积累诱导T细胞免疫抑制和Treg过度激活(图1)[87]。血清kyn/trp比值可能反映了抗pd -1免疫抵抗机制[88]。髓源性抑制细胞(Myeloid-derived suppressor cells, MDSCs)主要在肿瘤微环境中发挥免疫抑制作用[89]。一些炎症因子如TGF-β、IFN-γ和IL-6驱动着MDSCs的活化[90]。C-C基序趋化因子配体2 (CCL2)[91]和C-X-C基序趋化因子配体12 (CXCL12)[92]等趋化因子诱导MDSCs进入肿瘤部位。MDSCs通过上调PD-L1表达抑制肿瘤特异性T细胞的免疫应答(图1)[93]。
细胞外囊泡(EVs)
EVs是一组膜结合的载体,它们携带脂质、蛋白质和核酸[94]。通过内体途径向内出芽形成外泌体,从质膜外出芽形成微囊[95]。EVs与靶细胞结合并通过受体-配体相互作用启动信号转导或通过胞吞作用内化[96]。EVs介导癌细胞对化疗和放疗的敏感性,是一种很有前途的肿瘤诊断和预测标志物的液体活检策略[97,98]。免疫细胞间EVs的交换影响先天免疫和适应性免疫[99]。局部树突状细胞(dc)分泌- EVs可诱导T细胞活化[95]。EVs是微环境中连接肿瘤细胞与基质细胞之间沟通的关键成分[100]。通过从接受抗pd -1/PD-L1治疗的晚期NSCLC患者中提取EVs miRNAs进行测序分析,我们发现应答者与无应答者之间特异性miRNAs的浓度存在显著差异[101]。作为一种非侵入性液体活检,早期发现肿瘤源性EVs可能有助于预测抗PD -1/PD-L1治疗的疗效[102-104]。
外周血中的生物标记物
循环癌相关巨噬细胞样细胞(CAMLs)
肿瘤相关巨噬细胞(TAM)通过分泌VEGF、MMP、TNF-α等生长因子和细胞因子来促进恶性细胞的侵袭特性[105]。TAM和循环肿瘤细胞(CTC)通过淋巴屏障或毛细血管屏障迁移至血液循环,增强肿瘤侵袭和远处转移[106]。CAMLs是一种弥漫性TAM(图1),从各种癌症患者外周血中分离出CAMLs可能是肿瘤转移和新生血管形成的证据[107]。使用多重免疫染色的CellSieve系统定量CAMLs[108]。CAMLs≥50 μm为巨型CAMLs。CRT完成后CAMLs的大小与疾病进展和患者生存有关[109]。在抗PD-L1维持治疗前出现巨大CAMLs提示预后不良(中位PFS: 8个月,HR = 2.5, 95% CI: 1.1-5.8, p = 0.025;中位OS: 25个月,HR = 3.5, 95% CI: 1.3-9.6, p = 0.034)(表1)。CAMLs的肿瘤刺激作用可能限制抗PD-L1治疗的疗效[109]。、
PD - L1+非整倍体循环肿瘤内皮细胞(CTECs)
染色体非整倍性影响基因表达,决定肿瘤异质性,与肿瘤的进化密切相关[110-112]。CTECs,非整倍体CD31+循环肿瘤内皮细胞[113],来源于肿瘤组织中的非整倍体CD31+肿瘤内皮细胞,促进肿瘤血管生成[114,115]。免疫治疗后PD-L1+ CTECs发生形态学和核型改变[116]。Anti-PD-1能有效消除单倍体小CTECs,而相对增加多倍体大PD-L1+ CTECs[116]。PD-L1+ CTECs亚型患者抗pd -1治疗耐药。PD-L1+ CTECs患者的中位PFS为5个月(95% CI: 3.9-6.1个月)(表1),短于无PD-L1+ CTECs患者的中位PFS(8个月,95% CI: 4.9-11个月)。推测CTECs上的PD-L1与T细胞上的PD-1相互作用抑制了CD8+ T细胞的肿瘤特异性免疫攻击,影响免疫治疗的疗效(图1)[116]。
其他外周血细胞
在许多反映炎症的指标中,高中性粒细胞与淋巴细胞比值(NLR)预示着许多恶性肿瘤预后不良[117,118]。多项研究发现,NLR高的NSCLC患者对免疫检查点抑制剂(immune checkpoint inhibitors, ICIs)的反应率较低[119,120]。meta分析显示,术前NLR高的患者预后较差(PFS: HR = 1.44, 95%CI: 1.26-1.65, p < 0.001;OS: HR = 2.86, 95%CI: 2.11-3.87, p < 0.001)(表1)[119]。同样,另一项回顾性研究也验证了NLR对抗pd -1治疗的预测价值[120]。乳酸脱氢酶(LDH)是癌症相关炎症的一个指标[121]。根据LDH和NLR值将肺癌患者分为3组(良、零因子组;中间,1因素;穷,2因素)。与良好组相比,中间组和不良组更容易抵抗anti-PD-1/PD-L1治疗[121]。此外,NLR和LDH可能是预测irAEs的有用指标[122]。中性粒细胞与髓细胞表型高度相关,促进淋巴细胞耗竭[123]。T肿瘤浸润CD8+ T细胞与中性粒细胞(CD8/PMN)比值可以区分抗pd -1治疗的应答者[123]。联合中性粒细胞拮抗剂可改善免疫治疗结果[123]。同时,应答者NK细胞数量和活性均显著升高[124]。
肠道微生物群
肠道菌群与宿主存在共生关系[125]。微生物除了在胃肠道中起屏障作用外,还与菌群的免疫功能有关[126]。免疫细胞通过微生物蛋白和肿瘤抗原之间的交叉反应被激活[127]。DCs在肠外诱导活化的T细胞,识别肿瘤抗原并发挥抗肿瘤作用[127]。此外,微生物蛋白从肠道转移到血液循环,触发次级淋巴样器官的初始免疫,并诱导T细胞的激活。T细胞迁移到肿瘤部位并参与免疫监测(图1)[127]。微生物的组成可能会影响PD-1抑制剂的疗效[128]。一项研究表明,69%(11/16)和58%(23/40)的患者表现出部分反应或疾病稳定,而34%(15/44)的进展或死亡患者可检测到粪便粘虫[129]。通过16S核糖体RNA基因测序可以评估粪便标本的肠道菌群分布。腐肉毒杆菌、copri普雷沃氏菌和长双歧杆菌在有反应者中富集,未分类瘤胃球菌在无反应者中富集。微生物群多样性越高的患者,PFS明显越长(HR = 4.2, 95%CI: 1.42-12.3, p = 0.009)(表1)[130]。在不同的研究中,与临床获益相关的微生物群是不同的,这意味着饮食、宿主遗传、生活方式因素和人类物种之间的差异可能有助于肠道微生物群的多样性,并进一步影响ICIs的疗效[131,132]。累积抗生素(ATB)的应用会降低肠道菌群多样性,破坏微生物平衡[133,134],显著削弱PD-L1抑制剂的疗效,影响生存结局(中位PFS: 1.9个月,HR = 1.5, 95%CI: 1.0-2.2, p = 0.03;中位OS: 7.9个月,HR = 4.4, 95%CI: 2.6-7.7, p < 0.01[135]。有研究表明质子泵抑制剂(PPI)通过胃酸影响肠道菌群多样性[136]。第二阶段的数据白杨树和三期橡树试验表明,在人口anti-PD-L1疗法,患者作为或PPI较短的操作系统(HR = 1.20, 95%置信区间ci: 1.04 - -1.39)(表1),和应用程序相关的PPI明显短PFS (HR = 1.26, 95% ci: 1.10―-1.44)[137]。粪便微生物组移植(FMT)是一种很有前途的治疗方法,可以提高肠道菌群的多样性和免疫治疗的疗效[138,139]。
病人的临床特点
基因、激素等因素导致了男性和女性免疫反应的差异[140]。这种差异可能会影响男性和女性恶性肿瘤免疫治疗的疗效[140]。在一项meta分析中,通过比较抗pd -1/ PD-L1联合化疗和单纯化疗对男性和女性的影响,发现男性合并的OS-HRs为0.76 (95% CI: 0.66-0.87),女性合并的OS-HRs为0.48 (95% CI: 0.35-0.67)[141]。另一项荟萃分析显示,单抗pd -1组男性的综合OS-HRs为0.78 (95% CI: 0.60 - 1.00),女性为0.97 (95% CI: 0.79-1.19),而男性为0.76 (95% CI: 0.64-0.91),女性为0.44 (95% CI: 0.44)。0.25-0.76)的女性抗pd -1/PD-L1联合化疗[141](表1)。这意味着抗pd -1单药治疗可能对男性有更大的影响,而女性可能从抗pd -1/PD-L1联合化疗中获得更大的生存收益[141]。近80%的肺癌与吸烟有关。探索性分析显示,在接受抗pd -1治疗的患者中,目前和曾经吸烟的患者的总缓解率显著高于非吸烟者(36 vs. 26 vs. 14%)(表1)[142]。此外,吸烟年限的增加与抗pd -1治疗反应阳性相关[143]。通过公式(PS × BMI/LOT ×年龄)计算patras免疫治疗评分(PIOS),包括患者的行为状态(PS)、体重指数(BMI)、治疗线(LOT)和年龄。PIOS评分高的患者对抗pd -1治疗的反应最好(中位PFS: 15个月vs. 5个月,HR = 0.469, 95% CI: 0.295-0.747;中位OS: 32个月vs. 14个月,HR = 0.539, 95% CI: 0.317-0.918)(表1)[144]。
结论
抗pd -1/PD-L1治疗是一种很有前途的非小细胞肺癌治疗策略。然而,仍有大量患者难以从抗pd -1/ PD-L1治疗中获益。人们正在探索各种预测疗效的生物标志物。现阶段,PD-L1的表达是临床应用最广泛的生物标志物。TMB、TIL和新抗原与抗pd -1/PD-L1治疗效果显著相关。肠道菌群、炎症基因和失调的miRNA在抗肿瘤免疫调节中发挥重要作用。多种生物标志物的结合可以提高预测的稳健性,并指导癌症精准医学的实施。
